English
English Italiano
ItalianoCraniopharyngiomas are relatively rare tumors that arise in or near the pituitary gland.
Craniopharyngiomas make up 2.4 to 4 percent of all brain tumors, and are observed more frequently in children than in adults. While craniopharyngiomas are benign tumors, they may grow quickly and tend to recur after treatment.
The tumors usually are not discovered until they directly affect important structures around them, so they frequently are large when detected.
Symptoms
Symptoms of craniopharyngiomas are caused by the compression of adjacent structures as the tumor grows.
The tumors usually are located near critical structures, including the pituitary gland, hypothalamus, optic nerves, and important arteries.
If the tumor compresses the stalk of the pituitary gland or involves the gland itself, for example, it may result in
- pituitary hormone deficiency
that contributes to
- retarded growth
- obesity,
- a range of other hormonal problems
Similarly, visual deficits may result if the tumor affects the visual pathways or nerves.
Large tumors also will block the flow of cerebrospinal fluid, which bathes the brain and spinal cord, resulting in hydrocephalus, an increase in pressure within the brain that causes headache, nausea, and vomiting.
Diagnosis
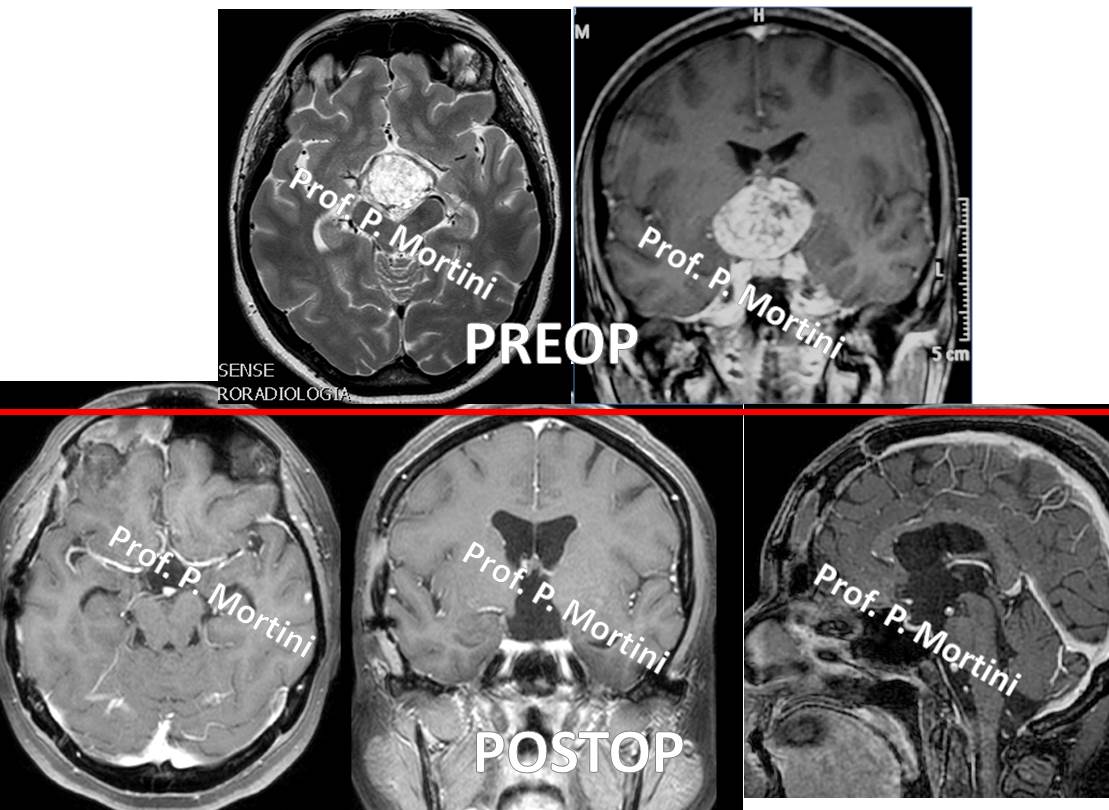
Imaging studies are the key component in the diagnosis of craniopharyngiomas. A contrast agent is administered intravenously so neurosurgeons can visualize the tumor against the normal brain in the background.
• Magnetic Resonance imaging (MRI)
• Computed Tomography (CT) scans also are used.
• In some cases, neurological surgeons may employ an MRI scan with frameless stereotactic guidance. For this study, a contrast MRI is performed after special markers (called fiducials) are placed on the child’s scalp. The fiducials are processed by a computer, which calculates the location of the tumor and creates a three-dimensional reconstruction. This image then is used at the time of surgery to help locate the tumor precisely, maximize tumor removal, and minimize injury to the surrounding brain.
• In children with craniopharyngiomas, ophthalmologic and endocrine evaluations also are advisable.
Treatment
The initial treatment for a craniopharyngioma usually is surgery to remove as much of the tumor as possible while preserving pituitary and neural function.
Unfortunately, because these tumors tend to adhere to nearby structures, neurosurgeons sometimes must make a choice between a total resection, which carries the risk of significant neurological and endocrine problems, and an incomplete resection, which does little to stop the tumor from recurring.
Even in cases of total resection, these tumors tend to return, so repeat MRI or CT scans are conducted for a period of time following initial treatment. Also, long-term hormone replacement therapy may be necessary after surgery.
Radiation therapy is used as a follow-up to surgery, especially in cases of less than total resection, but may have adverse effects on the development of young children. In addition, some tumors may be treated effectively with Stereotactic Radiosurgery, which involves the use of a highly focused beam of radiation to target the cancer cells specifically and leave the surrounding brain unaffected.